Challenge: Scaling protein docking consensus algorithms
The relationship between protein-protein interactions (PPIs) involved in mental illness has been established, yet most of these interactions lack experimental structures of the complexes formed by the interacting proteins. This gap necessitates the use of molecular docking simulation programs to predict these crucial structures.
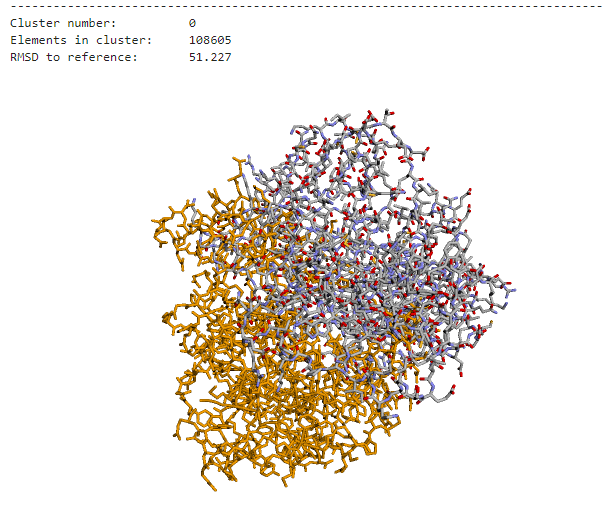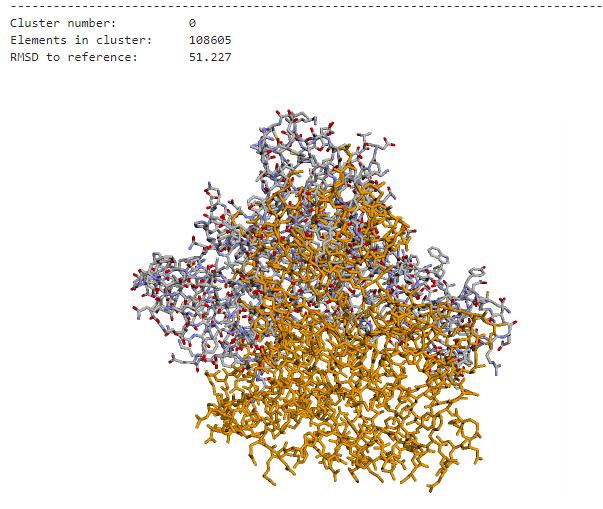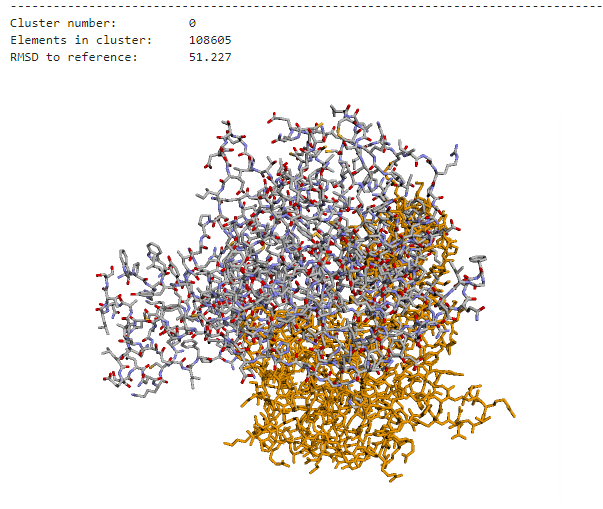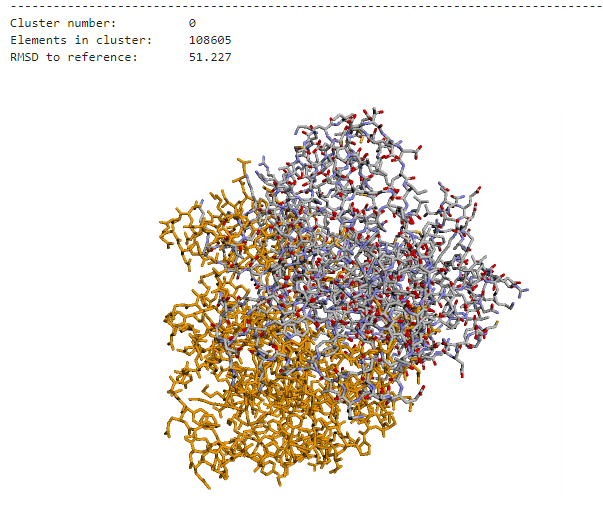
However, current docking programs struggle with accurately ranking the thousands of predicted structures they generate. The metrics used by these programs to classify predictions are not comparable, and existing consensus algorithms—while effective—are not scalable for handling large amounts of docking poses. This creates a significant bottleneck in computational biology research.
The challenge was clear: develop an efficient consensus algorithm that could process massive datasets of docking poses while maintaining clustering accuracy. Traditional approaches were simply too slow to handle the scale of modern computational biology requirements, taking hours to process datasets that needed to be analyzed in minutes.

